
ACE Alzheimer’s: An adjuvant strategy of treating Alzheimer’s disease with Vitamin A, C & E (ACE)
ABSTRACT
Alzheimer’s disease (AD) is a chronic and slowly progressing neurodegenerative disorder which has become a major concern with regards to health, worldwide. This disorder is characterised by progressive dementia and cognitive decline. Pathologically, AD is characterised by the presence of Aβ plaques and tau neurofibrils. However, literature has shown that oxidative stress is one of the most important risk factor behind the cause of AD. Oxidative stress often leads to production of Reactive Oxygen Species (ROS), which further increases structural and functional abnormalities in neurons of the brain, which subsequently, presents as dementia and cognitive decline.
In order, to curb the oxidative stress, antioxidants can be of great help. There have been many evidences that supports the use of antioxidants in the treatment for AD. Vitamins A, C and E are an example of antioxidants that can be used as adjuvants in the treatment of AD. This article will focus on current literature and will present forward the evidence based advantages of using Vitamin A, C and E as an adjuvant treatment for AD.
Keywords: Antioxidants, ACE, Adjuvant therapy.
INTRODUCTION
A clinical psychiatrist and neuroanatomist, Alois Alzheimer, reported “A peculiar severe disease process of the cerebral cortex” to the 37th Meeting of South-West German Psychiatrists in Tubingen, thus marking the discovery of one of the most interesting pathologies in medicine – Alzheimer’s disease. His invention was based on the observations in one his patient named Auguste D, suffering from profound memory loss, unfounded suspicions about her family, and additional worsening psychological changes. Her post mortem findings further revealed dramatic shrinkage of the brain and abnormal deposits in and encircling the nerve cells [1].
AD has proven to be a significant public health issue, as it consumes a major amount of heath budget in developed as well as developing countries. AD has become one of the leading causes of dementia in patients less than 65 years, other causes being Lewy body dementia (LBD), frontotemporal dementia (FTD), vascular dementia (VaD) and alcohol associated dementia [2].
United States alone has documented a $200 billion annual expenditure on patients affected by AD. Moreover, one person develops Alzheimer’s dementia every 68 seconds emphasizing the incidence of the disease [3]. Dementia can be defined as a chronic progressive disorder marked by memory deficits, personality changes, and impaired reasoning.
Results from population-based studies have shown a significant relationship between the certain risk factors and development of AD. Increased risk was shown with an increase in age, fewer years of education, and head trauma. Genetic factors do contribute to the early development of AD – increased risk with mutations on chromosome 21 (cases of down’s syndrome) as it carries the amyloid precursor protein, the presence of apolipoprotein E epsilon 4 allele and the presenilin 1 and 2 genes. The strongest factor identified till date are the apolipoprotein E genes located on chromosome 19 which exists in three forms – ε2, ε3, and ε4. ε2 has been found to reduce the risk, ε3 is found to be neutral whereas ε4 has been associated with a tremendous increase in risk as well as early development of symptoms (Figure 1) [4].
|
Chromosome |
Genes |
|
21 |
AMYLOID PRECURSOR PROTEIN |
|
19 |
APOLIPOPROTEIN E |
|
14 |
PRESENILIN 1 |
|
1 |
PRESENILIN 2 |
Figure 1. Genetic factors causing a risk to develop Alzheimer’s
Ad is difficult to differentiate from other causes of dementia like LBD, FTD and Vad [5]. It may present with dysfunction of various fields such as vision, touch – voluntary movements, personality deficits and judgemental disorders depending upon the area of the brain affected [6].The National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s disease and Related Disorders Association (NINCDS/ADRDA) has proposed a diagnostic criteria for differentiating between AD and other known causes of dementia. In compliance with NINCDS/ADRDA , AD is diagnosed if: (I) Cognitive functions decline progressively over a period of time including/ not including memory impairment or (Ia) Inability to understand language and verbal commands (aphasia); (Ib) Loss of ability to accomplish tasks due to incoordination of muscles (apraxia); (Ic) Failure to recognise previously known objects and loss of ability to use them(agnosia); (Id) Unable to plan, organise and execute daily chores; (II) All above mentioned under ‘I’ do get progressively deteriorated with time; (III) Other known causes of dementia as well as cognitive deterioration must be eliminated [6,7].
Neurofibrillatory tangles and extracellular amyloid plaques have been the initial histopathological findings associated with AD. Recently several other features have been recognised which include degeneration of neuronal synapses, aneuploidy and loss of neurons in the hippocampus. Despite the recent inventions, presence of extracellular amyloid plaques and intracellular NFT have been taken into account as the main histopathological criteria for establishment of AD [8]. Among all the different hypothesis, Aβ cascade has been the most accepted. Previously, a mutation in beta-Amyloid Precursor Protein (APP), which contributes to the normal function of neurons and cerebral development, was thought to be the sole culprit since the accumulation of Aβ proteins had lead to the pathogenesis of AD [9]. Eventually, mutated presenilin genes (both 1 and 2) have been discovered to play a role in the formation of Aβ pools [10]. But the exact mechanism underlying how Aβ aggregation contributes to the pathophysiology of AD largely remains unclear. Formerly, toxicity of neurons was believed to be caused by intracellular plaques. But recent data has suggested the role of intracellular Aβ proteins, which do not become sequestered into the extracellular plaques, as the toxic triggers stimulating the progression of AD [11]. Recently, it has also been shown that intracellular accumulation of Aβ proteins precedes the formation of extracellular Aβ protein plaques and NFT formation [12]. The role of intracellular Aβ protein in the progression of AD has also been demonstrated in recent experiments on transgenic mice. Results of these experiments indicate that increased deposits of Aβ proteins within the cells are associated with accelerated cell death [13].
Other important causative factors in the development of AD include oxidative stress and Reactive Oxygen Species (ROS) [14]. Susceptibility to oxidative damage is due to several factors which include relatively lower levels of antioxidants, significantly higher levels of polyunsaturated fatty acids, (these fatty acids rapidly fall prey to ROS), the presence of metallic ions and high oxygen utilisation [15]. Oxidation have been prove to be fatal for several constituents of the cells including carbohydrates, lipids, proteins, RNA and DNA [16]. Indirect mechanisms do play a vital role in the damaging process. Oxidation has been proven to accelerate the expression of inducible nitric oxide (iNOS) and accentuate the activity of neuronal NOS (nNOS). This leads to increased production of nitric oxide (NO). NO is known to interact with super oxide anions thus forming a highly reactive peroxynitrite anion. These transient molecules exerts their effects mainly on sulfhydryl groups of cells. [17]. The entire process has been depicted in figure 2.
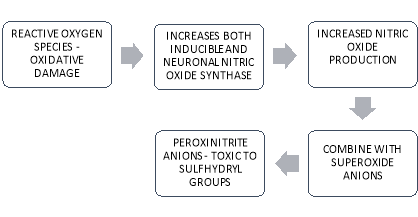
Figure 2. Nitric Oxide Pathogenesis
In addition to the indirect mechanisms, oxidative stress alters the protein structure. Impaired proteins are known to accelerate oxidative damage, thus proven to be interrelated. ROS causes the protein to be oxidised leading to a modified structure and causing them to be dimerized and aggregated [18]. Thus the oxidised protein which is both structurally and functionally abnormal gather as inclusions within the cytoplasm of the neurons, seen in the form of NFT (tau aggregates) and Aβ plaques [19]. Alternatively, Aβ plaques can also lead to the increased production of ROS. The entire process has been depicted in figure 3.
OXIDATION ALTERED PROTEIN STRUCTURE
CYTOPLASMIC INCLUSIONS DIMERISATION & AGGREGATION
Figure 3.Displaying Correlation between Oxidation and Protein Dimerization, thus forming a Vicious Cycle
Aβ (1-42) is an abundant species of Aβ proteins seen in AD [20]. Aβ (1-42) peptides is known for its toxicity which can be attributed to a residue of methionine at position 35 [21]. Oxidation of methionine contributes to the formation of methionine sulfoxide, which generally leads to irreversible oxidation and subsequently, forming methionine sulfone [22]. Methionine sulfoxide reductase (MSR) can even help the reduction of methionine sulfoxide into methionine [23]. However, the activity of MSR is also observed to be impaired in AD [24]. Methionine peroxide plays an important role in oxidative stress and toxicity caused by Aβ (1-42) peptides. The lone-pair of electrons present on the S atom of methionine undergoes oxidation of one atom and as a result, sulfuranyl radicals (MetS.+) are generated [21,25]. Sulfuranyl radicals are known to trigger the generation of other ROS like sulfoxides and superoxides by interacting with molecular oxygen [26].
The reason behind this intense oxidative damage could be attributed to the relative absence or decreased function of different antioxidant mechanisms of the body. Glutathione is one of the major antioxidant which can protect the brain tissues by causing detoxification of damaging ROS [27]. One of the main reasons of increase in oxidative stress in AD is the decreased glutathione levels in the brain [28]. The other members of the cellular antioxidant mechanism which plays a pivotal role includes Superoxide Dismutase (SOD) and Catalase (CAT). SOD is an antioxidant which is responsible for converting toxic superoxide ions into far less toxic hydrogen peroxide [29]. CAT evolves this reaction in to one step further and turns hydrogen peroxide into water [30].
Investigations have revealed that the levels of SOD and CAT decline in patients with AD [31]. Glutathione reductase (GR) and Glutathione peroxidase (GPx) represent the other crucial parts of the cellular defence mechanism which acts against oxidative stress. GPx is responsible for the metabolism of hydrogen peroxide and lipid hydroperoxides [32] and GR accelerates the reaction which helps in the regeneration of Glutathione (GSH) [33]. In total, the combination of an oxidative stress with above mentioned cellular defence mechanism against ROS, leads to the pathogenesis of AD. The pathogenesis of Alzheimer’s disease is mentioned in Figure 4.
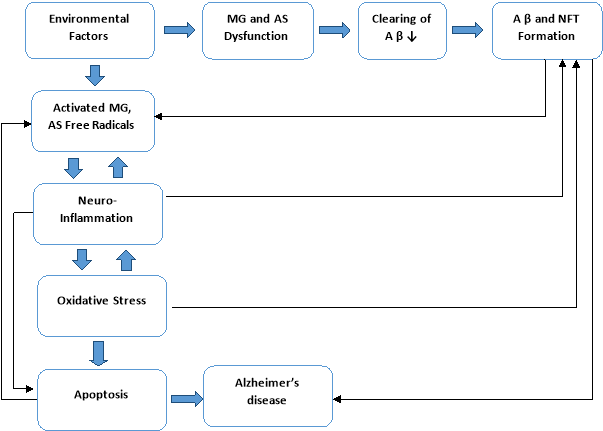
Figure 4. Pathogenesis of Alzheimer’s disease (MG : Microglia ; AS: Astrocyte; AP: Amyloid protein beta; NFT: Neurofibrillary tangles)
ACE ALZEIHMERS: VITAMIN A, C & E (ACE) THERAPY
ROLE OF VITAMIN A
Vitamin A and beta carotene have been shown to have multiple benefits for people suffering from AD. Various studies have found that patients suffering from AD have significantly lower levels of Vitamin A level and beta carotene in their CSF as well as blood [34]. The development of neurodegenerative disorders has shown to be influenced by Vitamin A and beta-carotene. Vitamin A plays an active role in neuronal development both in early life and in the adult nervous system. It protects and assists in the regeneration of neurons during recovery from neurodegeneration [35].
Inhibition of formation and destabilization of Aβ fibrils is an additional effect of Vitamin A and beta-carotene [35]. Since oligomerization of Aβ fibrils is an important mechanism contributing to neuronal toxicity in AD, Vitamin A supplementation has been shown to decrease the aggregation and oligomerization of Aβ40 and Aβ42 fibrils [36]. It has also been shown that Vitamin A and beta carotene decrease the decline of cognitive function in AD. Moreover, higher levels of these vitamins have been associated with better memory performance and spatial learning in these patients [34–36].
ROLE OF VITAMIN C
Various studies both in vivo and in vitro have shown to have significant effect in the brain due to decreased levels of vitamin C. Decreased plasma levels despite adequate intake in patients further confirmed the belief of protective effects of vitamin C in the spectrum of neurodegenerative diseases [37]. Hence, it can be proved that oxidative stress induces damage in AD and protection against this stress is offered to a certain degree by antioxidant vitamins. The progression of AD is altered by Vitamin C by interfering with various different aspects of pathology.
Numerous studies, both in-vivo and in vitro, have shown that Vitamin C can decrease oxidative stress. The structural progression of AD is prevented by Vitamin C by hindering the oligomerization of Aβ peptides [38]. Brain injury induces oxidative stress and reduces the level of antioxidants like vitamin C and SOD. Vitamin C supplementation improves the level of SOD, which consecutively helps to decrease oxidative stress and subsequent brain injury [39].
It has been suggested that even without additional supplementation, a normal intake of Vitamin C can have a neuroprotective effect in patients with AD. Cognitive decline in AD patients has shown to decrease is patients taking adequate Vitamin C [40]. In addition, results from a prospective observational study (n=4740) over a period of 3 years have shown that additional supplementation with antioxidant vitamins like vitamin C and E may be associated with both decreased incidence and prevalence of AD [41].
ROLE OF VITAMIN E
Vitamin E represents a cluster of 8 antioxidants composed of 4 tocotrienols and 4 tocopherols. It has been reported that there is a greater risk of neurodegenerative disorders like AD and Mild Cognitive Impairment (MCI) with lower plasma levels of vitamin E. Additionally, the level of vitamin E metabolic products (5-nitro-γ-tocopherol etc.) is shown to increase significantly in AD and MCI [42].
Deficiency of Vitamin E can lead to the damage and destruction of neurons and has been implicated in cases of cerebellar atrophy [43]. Vitamin E is a potent antioxidant which can delay the progression of AD at several levels. Increased oxidative stress induced by Aβ plaques is known to be a risk factor for neuronal death and ensuing brain injury in AD. Vitamin E behaves like a scavenger for these free radicals and therefore, is neuroprotective. [44].
Vitamin E also provides protection against AD via various other methods. For example, the 12-lipoxygenase pathway leads to glutamate-induced neuronal cell death by inflammation. Vitamin E can reduce this inflammation induced neuronal death [45]. Furthermore, consumption of vitamin E has been linked with the regeneration of SOD, levels of which are shown to decline in AD [39]. Among the different forms of vitamin E, the greatest degree of protection against AD is provided by α-tocopherols and γ-tocopherols [46].
A population-based cohort study of 5395 individuals was conducted to evaluate the efficacy of dietary supplementation of antioxidants to provide protection against AD. Among all the antioxidants used, results showed that the most significant degree of protection (p=0.02) against dementia and AD was provided by Vitamin E [47]. Moreover, supplementation of 30 International Units of alpha-tocopherols can act as a valuable adjuvant in the treatment of various neurodegenerative diseases, including AD [48].
Conclusion
Alzheimer’s disease represents one of the most significant age-related neurodegenerative disorders. Oxidative stress is one of the most important mechanisms involved in the development and progression of this condition. In order, to curb the oxidative stress, antioxidants can be of great help. The use of antioxidant vitamins A, C and E as adjuvant therapy for AD has always been given consideration. Thus, further clinical research is necessary to study the potential of these vitamins such that it can be integrated into clinical treatment to accelerate the recovery of patients afflicted by this disorder.
REFERENCES
- Alzheimer A, Uber eine eigenartige Erkrankung der Hirnrinde Allgemeine Zeits Psychiat Psychisch Gerichtlich Med 1907 64:146-48.
- Harvey RJ, Skelton-Robinson M, Rossor MN, The prevalence and causes of dementia in people under the age of 65 years J Neurol Neurosurg Psychiatry 2003 74:1206-09.
- Alzheimer’s Association2012 Alzheimer’s disease facts and figures Alzheimers Dement 2012 8:131-68.
- Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging Am J Epidemiol 2002 156:445-53.
- Karantzoulis S, Galvin JE, Distinguishing Alzheimer’s disease from other major forms of dementia Expert Rev Neurother 2011 11:1579-91.
- Castellani RJ, Rolston RK, Smith MA, Alzheimer Disease Dis Mon 2010 56:484-546.
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease Alzheimers Dement 2011 7:263-69.
- Swerdlow RH, Pathogenesis of Alzheimer’s disease Clin Interv Aging 2007 2:347-59.
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Beta-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity Cell 1995 81:525-31.
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Candidate gene for the chromosome 1 familial Alzheimer’s disease locus Science 1995 269:973-77.
- Lesne S, Kotilinek L, Amyloid plaques and amyloid-beta oligomers: An ongoing debate J Neurosci 2005 25:9319-20.
- Gouras GK, Tsai J, Naslund J, Vincent B, Vincent B, Edgar M, Intraneuronal Abeta42 accumulation in human brain Am J Pathol 2000 156:15-20.
- Bayer TA, Wirths O, Intracelluar accumulation of amylois beta- A predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease Front Aging Neurosci 2010 2:8
- Butterfield DA, Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review Free Radic Res 2002 36:1307-13.
- Butterfield DA, Castegna A, Lauderback CM, Drake J, Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death Neurobiol Aging 2002 23:655-64.
- Butterfield DA, Reed T, Newman SF, Sultana R, Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment Free Radic Biol Med 2007 43:658-77.
- Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS, Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide Chem Res Toxicol 1992 5:834-42.
- Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation J Neurochem 1995 65:2146-56.
- Butterfield DA, Kanski J, Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins Mech Ageing Dev 2001 122:945-62.
- Selkoe DJ, Alzheimer’s disease: genes, proteins, and therapy Physiol Rev 2001 81:741-66.
- Butterfield DA, Boyd-Kimball D, The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity Biochim Biophys Acta 2005 1703:149-56.
- Moskovitz J, Berlett BS, Poston JM, Stadtman ER, Methionine sulfoxidereductase in antioxidant defence Methods Enzymol 1999 300:239-44.
- Maher P, Redox control of neural function: background, mechanisms, and significance Antioxid Redox Signal 2006 8:1941-70.
- Gabbita SP, Aksenov MY, Lovell MA, Markesbery WR, Decrease in peptide methionine sulfoxidereductase in Alzheimer’s disease brain J Neurochem 1999 73:1660-66.
- Pogocki D, Schoneich C, Redox properties of Met(35) in neurotoxic beta-amyloid peptide. A molecular modeling study Chem Res Toxicol 2002 15:408-18.
- Milller B, Williams T, Schoneich C, Mechanism of sulfoxide formation through reaction of sulfur radical cation complexes with superoxide of hydroxide ion in oxygenated aqueous solution J Am Chem Soc 1996 118:11014-25.
- Dringen R, Gutterer JM, Hirrlinger J, Glutathione metabolism in brain: metabolic interaction between astrocytes and neurons in the defence against reactive oxygen species Eur J Biochem 2000 267:4912-16.
- Saharan S, Mandal PK, The emerging role of glutathione in Alzheimer’s disease J Alzheimers Dis 2014 40:519-29.
- Zelko IN, Mariani TJ, Folz RJ, Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression Free Radical Biol and Medi 2002 33:337-49.
- Chelikani P, Fita I, Loewen PC, Diversity of structures and properties among catalases Cell Mol Life Sci 2004 61:192-208.
- Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease Exp Neurol 1998 150:40-44.
- Arthur JR, The glutathione peroxidases Cell Mol Life Sci 2000 57:1825-35.
- Shigeoka S, Onishi T, Nakano Y, Kitaoka S, Characterisation and physiological function of glutathione reductase in Euglena gracilis z Biochem J 1987 242:511-15.
- Bourdel-Marchasson I, Delmas-Beauviex M-C, Peuchant E, Richard-Harston S, Decamps A, Reignier B, Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients Age and Ageing 2001 30:235-41.
- Ono K, Yamada M, Vitamin A and Alzheimer’s disease Geriatrics Gerontol Intl 2012 12:180-88.
- Takasaki J, Ono K, Yoshiike Y, Ikeda T, Morinaga A, Takashima A, Vitamin A has anti-oligomerization effects on amyloid-β in vitro J Alzheimer’s Dis 2011 27:271-80.
- Rivière S, Birlouez-Aragon I, Nourhashémi F, Vellas B, Low plasma vitamin C in Alzheimer patients despite an adequate diet Int J Geriatr Psychiatry 1998 13:749-54.
- Montilla-López P, Muoz-Águeda MC, FeijóoLópez M, Muñoz-Castañeda JR, Bujalance-Arenas I, Túnez-Fiñana I, Comparison of melatonin versus vitamin C on oxidative stress and antioxidant enzyme activity in Alzheimer’s disease induced by okadaic acid in neuroblastoma cells Eur J of Pharmacol 2002 451:237-43.
- Ishaq GM, Saidu Y, Bilbis LS, Muhammad SA, Jinjir N, Shehu BB, Effects of α-tocopherol and ascorbic acid in the severity and management of traumatic brain injury in albino rats J Neurosci Rural Pract 2013 4:292-97.
- Harrison FH, A critical review of Vitamin C for the prevention of age-related cognitive decline and Alzheimer’s disease J Alzheimers Dis 2012 29:711-26.
- Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, Tschanz JT, Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study Arch Neurol 2004 61:82-88.
- Mangialasche F, Xu W, Kivipelto M, Costanzi E, Ercolani S, Pigliautile M, Tocopherols and tocotrienols plasma levels are associated with cognitive impairment Neurobiol Aging 2012 33:2282-90.
- Aoki K, Washimi Y, Fujimori N, Maruyama K, Maruyama K, Yanagisawa N, Familial idiopathic vitamin E deficiency associated with cerebellar atrophy Rinsho Shinkeigaku 1990 30:966-71.
- Yatin SM, Varadarajan S, Butterfield DA, Vitamin E prevents Alzheimer’s amyloid β-peptide (1-42)-induced neuronal protein oxidation and reactive oxygen species production J Alzheimer’s Dis 2000 2:123-31.
- Khanna S, Parinandi NL, Kotha SR, Roy S, Rick C, Bibus D, Nanomolar vitamin E α-tocotrienol inhibits glutamate-induced activation of phospholipase A2 and causes neuroprotection J Neurochem 2010 112:1249-60.
- Morris MC, Evans DA, Tangney CC, Bienias JL, Wilson RS, Aggarwal NT, Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change Am J Clin Nutr 2005 81:508-14.
- Devore EE, Grodstein F, van Rooij FJ, Hofman A, Stampfer MJ, Witteman JC, Dietary antioxidants and long-term risk of dementia Arch Neurol 2010 67:819-25.
- Pham DQ, Plakogiannis R, Vitamin E supplementation in Alzheimer’s disease, Parkinson’s disease, tardive dyskinesia, and cataract: Part 2 Ann Pharmaco ther 2005 39:2065-72.
Cite This Work
To export a reference to this article please select a referencing style below:
Related Content
All TagsContent relating to: "alzheimers"
Alzheimer’s disease (AD), is a degenerative disorder that leads to memory loss and bodily functions and is the most common form of dementia.
Related Articles
