
Overview
First described by Owen in 1868, Lissencephaly refers to a “smooth brain”. A typical brain is characterized by sulci, the grooves, and gyri, the folds or ridges (Toy and Tintner, 2013). However, an individual affected by lissencephaly demonstrates agyria or pachygyria. Agyria or pachygyria refers to the absence or incomplete development of the gyri and gives the brain an abnormally smooth appearance (see figure 1 and 2). The disorder can range from complete lack of the folds, as in the case of agyria, or can have a milder presentation in which there are “two distinct layers of cerebral cortex” (Lin & Carson-Dewitt, 2017). Lissencephaly is part of a spectrum of brain malformations and may exist as an isolated abnormality or associated with other syndromes that is observed in infants.
Lissencephaly can be divided into two main categories: Type I or classical lissencephaly and Type II or “cobblestone” lissencephaly. In infants affected by Type I lissencephaly, the brain is distinguished by the smooth surface of the cerebral cortex as well as the distinct presence of a 4-layered cortex rather than the normal 6 layers. Other abnormalities of the brain may also be associated with Type 1, such as the malformation of the corpus callosum or the cerebellum. While in Type II, the surface of the brain appears to have a bumpy appearance and the cortex lacks any distinguishable layers. Type II is said to affect muscle function (Lin & Carson-Dewitt, 2017).
Causes of isolated lissencephaly can be attributed to various factors, such as viral infections or insufficient blood flow to the brain during development. It can also be attributed to certain genetic factors. Studies have also been done to determine whether mutations arising in particular genes attribute to the disorder. LIS1, RELN, TUBA1A, NDE1, KATNB1, CDK5, ARX and DCX have shown linkage to the presentation of isolated lissencephaly and of the above listed, LIS1 and DXC gene mutations have been studied most frequently. Various gene mutations and deletions are still being discovered when it comes to these malformations and how some may lead to syndromes with very distinct clinical presentations and findings on imaging (León, M. I., Ruiz & Gutiérrez, 2011).
As the brain is an integral part of the Central Nervous System and the CNS is a topic heavily covered in BSC 2085, an exploration of this particular brain malformation would prove beneficial in tying together concepts covered. Despite the malformation being centralized on the brain, the presence of a disorder in an individual can cause problems in various other body systems and is an excellent example of seeing how all body systems relate to one another and the body functions, as well as how problems that can arise in embryonic formation can have a profound impact on overall development.
Diagnosis, Signs, and Symptoms
Though the signs outnumber the symptoms, a patient with lissencephaly may often complain of difficulty feeding or difficulty swallowing. Babies with the disorder may appear normal when first born but as they grow, the signs of the disease begin to manifest. An infant with lissencephaly can display severe developmental delay as well as failure to thrive at several months of age and also begin to suffer from seizures. The seizures that accompany the disorder begin in the first few months of life and are often difficult to treat. Some infants may also display microcephaly, an abnormally small head size. Cerebral palsy and decreased muscle tone or hypotonia may also develop later in life. Hydrocephalus, the presence of fluid in ventricles contributing to increased intracranial pressure, may also occur in some individuals.
Those with a milder form of lissencephaly may not show symptoms until later in early childhood and may only have mild developmental delay and seizures (Norman, Roberts, Sirois & Tremblay, 1976).
Diagnosis of lissencephaly is often confirmed through clinical evaluation and brain imaging studies, such as computerized tomography (CT) scanning or magnetic resonance imaging (MRI). CT or MRI imaging obtained in infancy shows poor development of cerebral sulci (refer to figure 4). The Sylvian fissure has a superficial appearance and maintains a “rounded morphology due to delayed insular operculation”, which should already be evident in appearance after the 25th gestational week (León, M. I., Ruiz & Gutiérrez, 2011). Ultrasounds (US) performed at the prenatal stage can also suggest the presentation of lissencephaly type 1 (refer to figure 3) and if there is family history of the disorder accompanying suspicious prenatal US findings, the condition may be confirmed though specialized testing during pregnancy, i.e. cell-free fetal DNA, amniocentesis or chorionic villus sampling (CVS). In these tests, samples of amniotic fluid or chorionic villi can be tested to see if there is a causative mutation on chromosome 17 (Lin & Carson-Dewitt, 2017).
Treatment
There is no definitive treatment for lissencephaly and any treatments are purely symptomatic. Treatment is geared towards the symptoms that present in each individual and as such, it’s highly specialized per person. The course of treatment for a patient may also need the combined efforts of a specialist team to manage various aspects of the disorder, i.e. pediatricians, neurologists, and any other health care professionals.
To assist with poor feeding and failure to thrive, patients may often require the placements of gastrostomy tubes and be started on enteral feedings and improve nutrient intake. Seizures may also be controlled through the administration of various anticonvulsants, the dosage and type determined by neurologist closely following patient treatment. Individuals with hydrocephalus may also undergo shunting in order to drain the fluid and help relieve intracranial pressure (Lin & Carson-Dewitt, 2017).
Prognosis
There is no cure available for those with lissencephaly. Due to the varying severity of the disease, the prognosis of children affected greatly varies. Life expectancy, however, remains short and most children afflicted with the disorder will die before the ae of 10. Aspiration and respiratory disease or infections are the most common cause of illness—or death. Children with the disease will have developmental delay, though the degree is dependent on the severity of the disease. Some children can roll over and sit as well smile and reach for objects, however, most remain in a 3-5 month developmental level.
In the past, life expectancy was around 2 years old. But with the advent of better seizure control as well as treatments for respiratory problems as well as nutritional deficiencies, the life expectancy has grown (Lin & Carson-Dewitt, 2017).
Appendix
Figures 1-4 were obtained from Learning Pediatric Imaging: 100 Cases

Figure 1

Figure 2
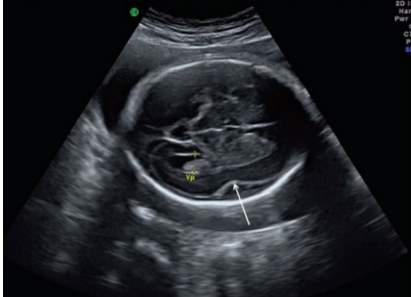
Figure 3

Figure 4
Bibliography
- León, M. I., Ruiz, L. C., & Gutiérrez, J. E. (2011). Learning pediatric imaging 100 essential cases. Heidelberg: Springer.
- Lin, P. T., MS, & Carson-Dewitt, R., MD. (2017). Lissencephaly. In D. S. Hiam (Ed.), The Gale Encyclopedia of Neurological Disorders (3rd ed., Vol. 2, pp. 711-714). Farmington Hills, MI: Gale. Retrieved from http://link.galegroup.com.ezproxy.fiu.edu/apps/doc/CX3662900225/GVRL?u=miam11506&sid=GVRL&xid=a37332b4
- Lissencephaly. (n.d.). Retrieved from https://rarediseases.org/rare-diseases/lissencephaly/#references
- Norman, M. G., Roberts, M., Sirois, J., & Tremblay, L. J. (1976, February). Lissencephaly. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/175907
- Toy, E. C., Simpson, E., & Tintner, R. (2013). Case files. New York: McGraw-Hill Medical.
Cite This Work
To export a reference to this article please select a referencing style below:
Related Content
All TagsContent relating to: "diagnosis"
Once a patient has been diagnosed, a plan of care should be actioned to include further diagnostic testing, medications, referrals, and follow-up care. Patient education should also be provided regarding diagnosis, exercise, diet, medicines, and warning signs of medication and diagnoses.
Related Articles
