
HIV Reverse Transcriptase: Molecular Mechanisms of Inhibition and Resistance
Structure of HIV
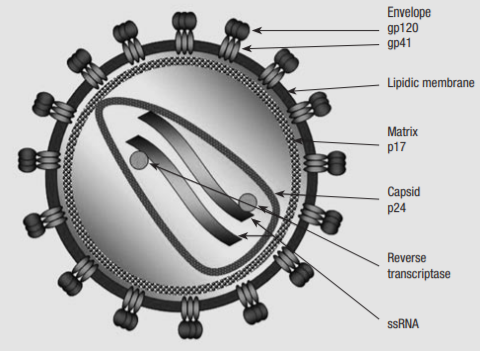
HIV is a lentivirus of the family retroviridae, which typically shows a chronic course of infection, with long clinical latency and persistent viral replication, with nervous system involvement. The structure of HIV differs to other lentiviruses. It is spherical, 100-120nm in diameter, consisting of a lipid bilayer taken from its host cell and a conical nucleocapsid comprised of p24 which contains the viral genome, protease (PR), reverse transcriptase (RT), integrase (IN), and various accessory proteins and cellular factors. The HIV genome is transmitted as two identical copies of 9.2kb single-stranded positive sense RNA.
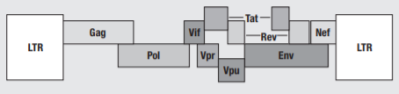
Figure 1 Organisation of the HIV-1 genome (1)
Figure 2 Structure of the HIV-1 particle (1)
The retroviral genome is identified by the structural genes Gag, Pol and Env. Gag codes viral core proteins matrix (MA), capsid (CA) and nucleocapsid (NC). The Pol gene is crucial to viral replication, as it encodes the enzymes which bring about the HIV replication cycle – RT converts viral RNA to DNA, IN integrates the viral DNA into the host chromosomal DNA, and PR cleaves the Gag and Pol precursors into their own parts. Env codes for gp120 and gp41, a surface and transmembrane protein respectively, which make up the envelope glycoproteins that bind to CXCR-4 and CCR-5 CD4+ coreceptors, initiating the infection by gaining entry to the host cell. The Env glycoprotein is the only protein on the surface of HIV-1, making it a key target for vaccine development. However it is also one of nature’s most heavily glycosylated proteins, which shields many immunogenic sites from detection.(2)
HIV-1 is characterised by other regulatory genes specific to it, which have key roles in modulating viral replication. These are summarised in Table 1.
|
Class |
Gene name |
Primary protein products |
Function |
|
Essential regulatory proteins |
tat |
Tat |
Expressed in early infection and promotes expression of HIV genes |
|
|
rev |
Rev |
Binds to viral genome and transports viral RNA from cytosol to nucleus during replication |
|
Accessory regulatory proteins |
nef |
Nef |
Cellular signalling transduction and downregulation of CD4+ receptors on cell surface membrane to allow viral budding |
|
vpr |
Vpr |
Nuclear import of preintegration complex, causes host cell to arrest in cell cycle at G2 to activate DNA repair mechanism to aid integration of viral DNA |
|
|
vif |
Vif |
Enhances infectivity of progeny virus particles |
|
|
vpu |
Vpu |
Involved in CD4 degredation involving ubiquitin proteasome pathway, and correct release of virions from infected cells |
Table 1 Regulatory proteins encoded by HIV-1 genome (3)
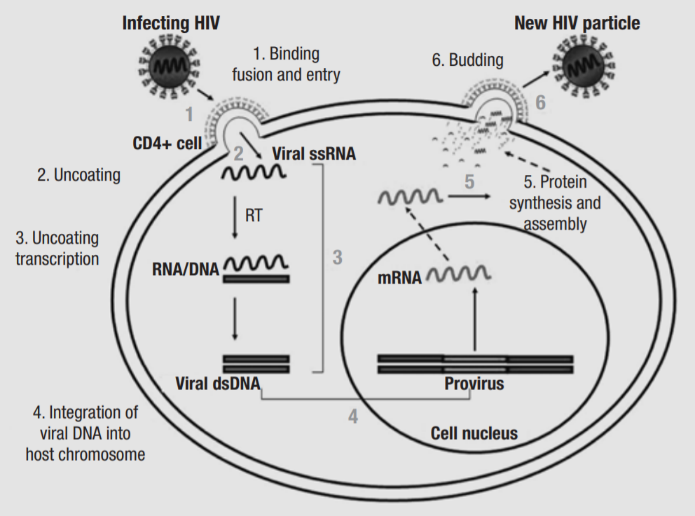
HIV-1 Replication
Figure 3 HIV replication cycle (1)
The replication cycle of HIV-1 can be summarised in six steps: binding and entry, uncoating, reverse transcription, provirus integration, virus protein synthesis, and budding. The process begins with the attachment of virions to the cell surface via interactions between gp120 and the CCR5 and CXCR4 coreceptors of CD4, and gp41 contains a fusogenic hydrophobic peptide at the amino terminus, which is critical for the fusion of viral and cell membranes (4). Once the membranes are fused, the nucleocapsid is released into the cytoplasm. Uncoating involve several cellular factors, as well as viral proteins MA, Nef and Vif. Next, HIV-1 RT begins the conversion of single-stranded viral RNA into the double-stranded DNA provirus, a process which will be explored in more detail later. The accuracy of RT is influenced by APOBEC3G, and Vif counters its antiretroviral effects by reducing its expression and stopping it from being incorporated into progeny virions (5). Vpr directs the preintegration complex to dock to the nuclear membrane and enters through the nuclear pore (6). The provirus is integrated into the host cell DNA by IN, and the first round of proviral transcription and translation of the Long Terminal Repeats produce basal amounts of Tat, Rev and Nef. After the LTRs, the next sequence transcribed is the Trans-Activation Responsive element, which forms a loop structure and enables Tat to bind, which is a potent transcriptional activator, enhancing the elongation ability of RNA polymerase II. Rev binds to the Rev Responsive Element which is present in singly-spliced and non-spliced viral RNA and facilitates transport of RNA from the nucleus to the cytoplasm.
The final stage of the replication cycle is the assembly of the new HIV-1 virions, which starts at the plasma membrane of the host cell. env is translated into precursor polyprotein gp160, which is glycosylated in the endoplasmic reticulum, then transported to the Golgi apparatus, where furin cleaves gp160 into gp41 and gp120. These glycoproteins are then transported to the plasma membrane, to which they are anchored. The gag-pol gene is translated into Gag (p55) and Gag-Pol (p160) polyproteins, which also associate to the plasma membrane with HIV genomic RNA as the virion begins to bud from the host cell. Budding triggers the activation of PR, which cleaves the Gag and Gag-Pol polyproteins, releasing structural proteins and enzymes, which undergo further interactions – CA and NC go on to form the nucleocapsid, and MA remains associated to the viral envelope.
Structure and Function of Reverse Transcriptase
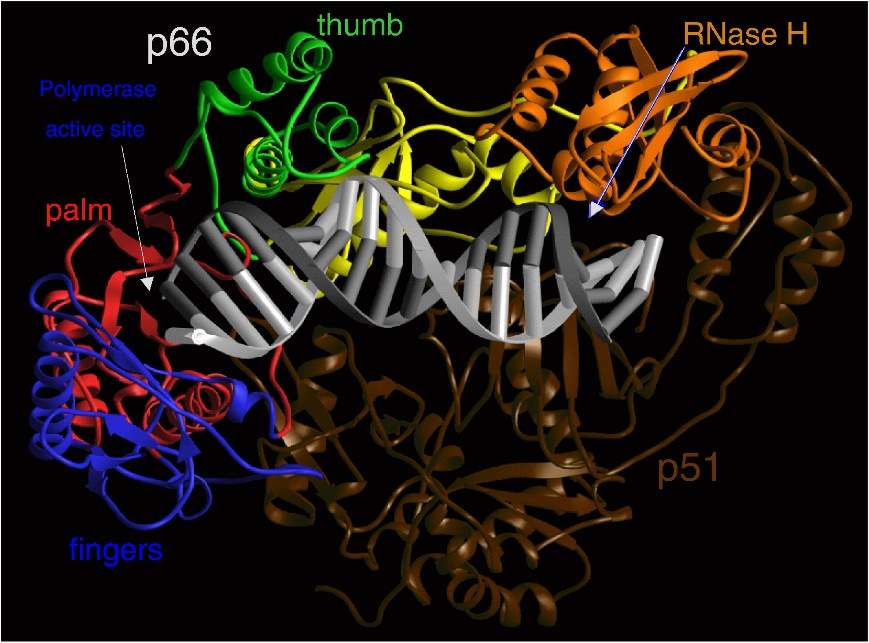
HIV RT is an asymmetric heterodimer made of two related subunits – p66 and p51. These subunits are derived from the cleavage of Gag and Gag-Pol polyproteins as mentioned previously. p66, the larger of the two subunits, contains the active site for both enzymatic parts of RT – the polymerase and Ribonuclease H (RNase H), while p51 has a structural role.
Figure 4 Ribbon representation of HIV-1 RT in a complex with nucleic acid. The fingers, palm, thumb, connection, and RNase H subdomains of the p66 subunit are shown in blue, red, green, yellow, and orange, respectively. The p51 subunit is shown in dark brown. The template and primer DNA strands are shown in light gray and dark gray, respectively. (7)
The polymerase domain of p66 is comprised of four subdomains: fingers, palm, thumb and connection. p51 also folds into the same subdomains but the positions of them relative to each other differ in p51 and p66 (Fig. 4). The nucleic-acid binding cleft is mainly formed by the fingers, palm, thumb, connection, and RNase H subdomains of p66, while the connection and thumb subdomains of p51 form the flood of the cleft. The binding cleft is constructed to bring the nucleic acid into contact with both the polymerase and RNase H active sites. The polymerase active site contains three catalytic carboxylates in the p66 palm subdomain that are required for catalysis, and there are several other highly conserved residues that make up the dNTP-binding site that interact with different functional groups. For example, the residue Q151 interacts directly with the 3’-OH of  the incoming dNTP, while R72 and K65 are involved with binding the β- and γ-phosphates (8).
the incoming dNTP, while R72 and K65 are involved with binding the β- and γ-phosphates (8).
Polymerase and RNase H cooperate to convert viral RNA into double-stranded DNA. RT requires both a primer and a template for DNA synthesis. In HIV-1, the primer is a host tRNAlys3. At the 5’ end of the viral genome, there is an 18-nucleotide long sequence called the Primer Binding Site (PBS) that is complementary to the 18 nucleotides at the 3’ end of tRNAlys3. tRNAlys3 hybridises to the PBS, and RT begins the synthesis of the complementary minus-strand DNA sequence for U5 and R (a direct repeat which is found at both ends of the viral RNA molecule. RNase H then degrades U5 and R at the 5’ end of the plus-strand RNA. The ‘first jump’ occurs, where the primer ‘jumps’ to the 3’ end, and the new DNA R region hybridises with the plus-strand RNA R region. The complementary minus-strand DNA is now extended by the polymerase, while RNase H degrades most of the viral RNA, leaving only the PP sequence. Plus-strand DNA synthesis begins, from the PP sequence to the primer, after which the second jump occurs. RNase H removes the tRNA primer, and the PBS of the plus-strand DNA hybridises with the PBS of the minus-strand DNA. Both the minus- and plus-strands of DNA are extended, creating a complete double-stranded DNA copy of the original viral genome.
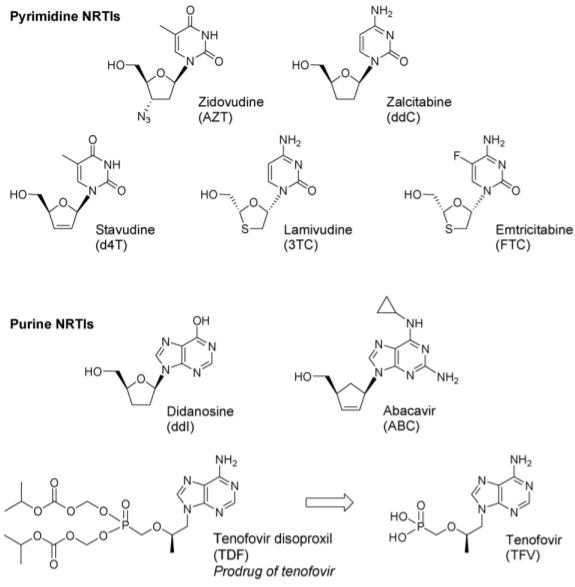
Figure 1 Structures of approved NRTIs (2)
AZT is a Nucleoside Reverse Transcriptase Inhibitor (NRTI), the first of six classes of antiretroviral drugs. NRTIs are the foundation of current HAART. According to NICE guidelines, treatment of HIV-1 infection is started with 2 nucleoside reverse transcriptase inhibitors, plus: a non-nucleoside reverse transcriptase inhibitor, or a boosted protease inhibitor, or an integrase inhibitor. The treatments of choice contain Tenofovir Disoproxil and Emtricitabine with a combination of various other antiretrovirals. Other regimens contain Abacavir and Lamivudine, with other protease and integrase inhibitors (3). Combination pills such as Combivir (AZT/3TC), Trizivir (AZT/ABC/3TC), and Truvada (TDF/FTC) are used to decrease the pill burden on patients, as these medications are lifelong.
NRTI Mechanism of Action
Nucleoside Reverse Transcriptase Inhibitors (NRTIs) are analogues of naturally occurring deoxynucleosides needed for the synthesis of viral DNA. In their parent form, they are inactive, and rely on host cell kinases and phosphotransferases for successive phosphorylation to deoxynucleotide triphosphate (dNTP) analogues which have inhibitory abilities. The NRTI dNTPs then compete with endogenous dNTPs to be incorporated into the viral DNA chain, acting as competitive substrate inhibitors. However, the key difference between the endogenous dNTPs and the NRTIs is that the NRTIs lack a 3’-hydroxyl group, and so once incorporated into the DNA chain, the incoming dNTP cannot form the next 5’-3’ phosphodiester bond needed to continue DNA synthesis. In this way, the NRTI acts as a chain terminator, forming a stable dead-end complex (4). There are many considerations that need to be made when designing an antiretroviral, as many factors such as efficiency of conversion to active metabolite and stability of the NRTI and the active dNTP form help determine blood concentrations of the drug required for efficacy. The intracellular retention of NRTI dNTPs allows for more constant viral inhibition. It has been proposed that the long intracellular half-lives of TFV-DP and FTC-TP contribute to the long-term clinical efficacy of the combination of TDF, FTC, and Efavirenz (5). Though all NRTIs are analogues of natural deoxynucleosides, not all NRTI are good kinase substrates, and phosphorylation can become a rate limiting step. Efficient intracellular phosphorylation of NRTI to NRTI-triphosphate (TP) is a vital requirement for the efficacy of an NRTI drug. For some NRTIs, like AZTTP, the addition of the second and third phosphate may be the rate limiting step, but for others it may be the addition of the first. Therefore, it can be helpful to have Nucleotide Reverse Transcriptase Inhibitors (NtRTIs), e.g. Tenofovir, which have the same antiviral effect as NRTIs, the only difference being that the drug molecule contains the first phosphate, which is called a prodrug. Prodrugs are also modified with hydrophobic groups to allow entry to the cell, which are removed upon entry (6)(7). NRTIs do not block the activity of RT, however, mutations in the dNTP binding cleft of RT can allow the enzyme to discriminate between NRTI dNTPs and endogenous dNTPs, leading to resistance.
NRTI Mechanisms of Resistance
There are two basic mechanisms of NRTI resistance: exclusion and excision. Exclusion is driven by enhanced recognition and discrimination of NRTI-dNTPs at the time of incorporation into the viral DNA. Excision involves selective removal of the NRTI-dNTP from the end of the viral DNA chain after incorporation.
Exclusion
All of the amino acids involved in the exclusion mechanism of resistance are located in the fingers and palm subdomains of RT – both positions which would affect the binding of an incoming dNTP (7). The M184V/I mutations give an example of the exclusion mechanism. With relation to Cytidine analogues 3TC (Lamivudine) and FTC (Emtricitabine), resistant viruses are selected with mutations at amino acid 184 of reverse transcriptase. M184 is a part of the dNTP binding site of HIV-1 RT, and structural studies propose that the way in which the mutation causes resistance involves steric hinderance. The stereochemical form of 3TC and FTC used in HIV-1 treatment are the opposite enantiomers to their normal dNTPs, and they also have their ribose ring replaces with an oxathiolane ring. Both the introduction of sulphur and the use of the opposite enantiomer causes the section of the oxathiolane ring containing the sulphur atom to project much further that the normal ribose ring, creating a possibility of steric hinderance. Since the wild-type enzyme readily incorporates 3TCTP and FTCTP, there can be no significant steric hinderance with M184. However, models generated based on a ternary complex comprising of the wild-type RT, double-stranded DNA and a bound dNTP indicate that a β-branched amino acid at position 184 would impede the ability of 3TCTP and FTCTP to bind in the correct configuration at the polymerase active site (8).
Q151 interacts with the 3’-OH of a normal dNTP. The Q151M mutation appears to cause changes in the hydrogen bonding network between the deoxyribose of the incoming dNTP and RT, which allows RT to differentiate between dNTPs with a 3’-OH and the NRTI dNTPs without. Q151M has four associated mutations: A62V, V75I, F77L, and F116Y, and together the mutations are referred to as the Q151M complex (Q151Mc). Q151Mc causes resistance to nearly all NRTIs except 3TC/FTC and TDF, and the viral strains carrying the mutations are termed multidrug-resistant (MDR) viruses (9)(10).
Excision
The excision mechanism involves the removal of the incorporated chain-terminating NRTI dNTP from the viral DNA strand by pyrophosphorolysis. The first example of NRTI resistance was resistance to AZT, and the molecular mechanism took more than ten years to be understood, partly because it was unexpected. A set of RT mutations (M41L, D67N, K70R, L210W, T215Y, and K219Q), known as thymidine analogue mutations (TAMs) or AZT-resistance mutations (AZTr), emerged in patients as distinctive mutations leading to AZT resistance. An AZT-resistant RT can incorporate AZTTP as effectively as a wild-type RT, but they have an augmented ability to excise the incorporated AZTMP from the 5’ end of the strand. The mechanism involved is ATP-dependant excision: a reversal of the normal process of polymerisation, where ATP is the pyrophosphate donor. This ‘unblocks’ the DNA chain, allowing it to be extended, and replication to continue (11). ATP-dependent excision, generated AZTppppA as the reaction product. Crystal structures of RT-DNA-AZTppppA and related complexes show that the mutations help create an ATP-binding pocket adjacent to the dNTP-binding cleft. The T215Y mutated aromatic side chain stacks with the base while the K70R mutated side chain forms polar interactions with the α–phosphate and 3′-OH of ATP (12).
Currently there are three generations of approved Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs): first, Nevirapine and Delavirdine, second, Efavirenz, and third, Etravirine and Rilpivirine.
NNRTI Mechanism of Action
NNRTIs have a high specificity, and only inhibit HIV-1, in comparison to NRTIs, which are broad spectrum antiretrovirals. When NNRTIs bind to RT at the NNRTI-binding pocket (NNIBP) in the palm subdomain of p66, at the base of the β4–β7–β8 sheet, approximately 10Å from the polymerase active site. The pocket itself is hydrophobic, lined with aromatic and hydrophilic amino acids. Structural studies show that the pocket is not visible when an NNRTI is not bound, but is created by the conformational change generated by the binding of an NNRTI, rotating the side chains of Y181 and Y188 towards the catalytic site. This causes a concomitant shift of 2Å in the β4–β7–β8 sheet and three catalytic aspartic acid residues. This structural change locks the fingers and thumb subdomains of p66 in a hyperextended configuration (1)(2). This distortion of the primer grip affects the positioning of the primer terminus in the polymerase active site. The hinge motion between the palm and thumb domains is critical for translocation of RT along the nucleic acid after each dNTP incorporation (3). Experiments were carried out attempting to form an RT-DNA-Nevirapine-AZTTP complex, but the only structure yielded was an RT-DNA-Nevirapine complex, with no well-defined binding of AZTTP in the polymerase active site. This suggested that when NNRTIs are bound to RT, dNTPs may enter the binding cleft, interact with RT and even cause conformational changes in RT e.g. closing the fingers, but catalytic formation of a -RT-DNA-dNTP complex would not occur (4).
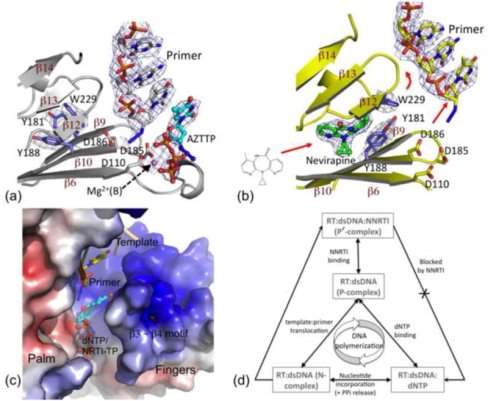
Figure 1 Structural basis for the inhibition of DNA polymerisation by an NNRTI. (a) Structure of RT-DNA-AZTTP ternary complex obtained by soaking AZTTP into crystals of RT-DNA complex. (b) Soaking of nevirapine into the crystal created the NNRTI pocket, repositioning the “primer grip” (on the β12–β13–β14 sheet) that moved the primer terminus away from the polymerase active site. (c) Electrostatic potential surface of RT bound to DNA and nevirapine. The crystallization experiments and structures showed an open dNTP-binding cleft into which dNTPs/NRTI-TPs can enter; however, structural aberration by NNRTI binding did not allow a dNTP to chelate metals and form base-pairing and base-stacking interactions. (d) These structural constraints preclude the formation of an RT-DNA-dNTP polymerase competent (P) complex, rather forms a non-productive (P′) complex in the presence of an NNRTI – a structural basis for NNRTI inhibition (4)
NNRTI Mechanisms of Resistance
Resistance mutations in NNRTIs usually occur in the NNIBP. K103N and Y181C are the most commonly observed mutations in patients. Others include L100I, K101E, V106A, V179D, Y188L, G190A, and P236L. These mutations can occur singly or in combination. High levels of resistance to NNRTIs are normally a consequence of monotherapy with these medications, particularly with first generation NNRTIs. Third generation NNRTIs are effective against strains with common single or double mutations, but strains with multiple mutations can cause substantial levels of resistance. There are three main resistance mechanisms: loss/change of key hydrophobic interactions, steric hinderance, and pocket entrance mutations.
Loss/change of key hydrophobic interactions
Mutations of key residues (Y181C, Y188L, F227L) in the hydrophobic core cause significant resistance due to the loss of interactions between the NNRTI and aromatic ring. This mutation is highly resistant to first generation NNRTIs as they are relatively rigid. However, second generation drugs are designed with ‘strategic flexibility,’ which allows them to have compensatory interactions with mutated RTs that are resistant to first generation drugs. This flexibility in the binding has been termed ‘wiggling and jiggling’ and it allowed NNRTIs to adapt to changes caused by resistance mutations in the NNIBP (5).
Steric hinderance
L100 and G190 are in the central region of the NNIBP. An L100I mutation changes the shape of the pocket, as the amino acid changes from being γ-branched to β-branched, whilst G190A mutations create a bulge. For example, HBY 097 (a compound that underwent pre-clinical evaluation as a potential NNRTI) can effectively inhibit RT with the Y181C mutation, but not those with G190A. Looking at the crystal structures of the RT-HBY 097 complex, the large and rigid quinoxaline ring of HBY 097 is near the β6-β10-β9 sheet containing G190. The mutation causes the Cβ atom of A190 to sterically clash with the quinoxaline ring, reducing the binding of HBY 097 (6).
Pocket entrance mutations
The K103N and K101E mutations frequently cause resistance to first generation NNRTIs. In fact, the K103N mutation causes a near consistent level of cross-resistance to most NNRTIs. The residues are located at the edge of the entrance to the NNIBP, and the mutation causes interference with NNRTI entry into the pocket. Second generation NNRTIs were designed to get around this, for example DAPY NNRTIs, e.g. Etravirine, are still able to inhibit mutant K103N RTs because they interact with the residue side chain – a hydrogen bond between Y188 phenoxyl group and the N103 side chain forms, which is not present in wild-type RT. This enhances binding (7) (8).
To conclude, NNRTI-resistance mutations seem to influence NNRTI binding directly, by changing the size, shape, and polarity of different parts of the NNIBP or, indirectly, by altering access to the pocket.
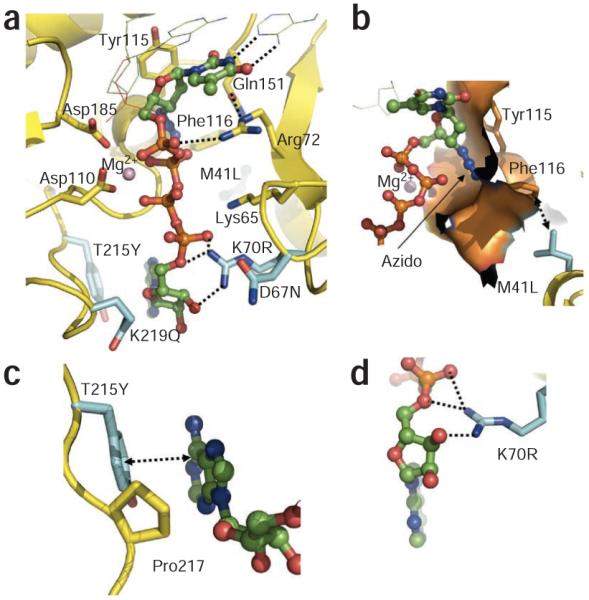
Figure 2 Binding of AZTppppA’ to AZTr HIV-1 reverse transcriptase. (a) Interactions of AZTppppA’ (carbons, green; phosphates, orange) with the mutated residues (cyan) and with the active site residues; hydrogen bonds, dotted lines; nucleic acid, thin lines. (b) Molecular surface representing the azido-binding cleft at the N site. (c) π-π stacking between Tyr215 and the adenine ring, and the hydrophobic interaction between Pro217 and the ribose ring stabilize the binding of ATP’ to AZTr reverse transcriptase. (d) Primary mutation K70R enhances ATP’ binding by interacting with the ribose ring and α phosphate. Structure images were made using PyMOL (http://www.pymol.org/).
Cite This Work
To export a reference to this article please select a referencing style below:
Related Content
All TagsContent relating to: "HIV"
The human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) pandemic is one of the most serious contemporary sexual health related issue affecting the human race today. By the end of 2009, it was approximated that 34 million people were living with the HIV virus and deaths related to AIDS were about 1.8 million people.
Related Articles
